Crystal structure and cryo-electron microscopy structure are the structural foundation for computer-aided drug design. However, these experimental structures can only give static information, while the recognition of ligand-protein and protein-protein interactions is a dynamic process, which undergoes many important intermediate states. Future drug design includes not only the ligand-receptor binding states observed in experiments but also various intermediate states in the whole ligand-protein recognition pathway. Intermediate states can provide a more detailed supplementary structure and offer new insights into drug design. The recognition of ligand-protein and protein-protein occurs on microsecond (μs), millisecond (ms), or an even longer time scale, and it is difficult to simulate large-scale conformational changes within a reasonable time by conventional molecular dynamics (cMD) methods. Therefore, it is necessary to develop accelerated sampling methods to improve simulation efficiency and capture more dynamic information in a fair simulation period.
Recently, the team led by Professor Lin Jianping and Associate Professor Li Dongmei of the College of Pharmacy of Nankai University and the State Key Laboratory of Medicinal Chemical Biology published a paper titled "The Full Activation Mechanism of the Adenosine A1 Receptor Revealed by GaMD and Su-GaMD Simulations" on Proceedings of the National Academy of Sciences of the United States of America (PNAS). The research team developed a new MD method, Su-GaMD, and revealed the full activation mechanism of adenosine (Ado) A1 receptor (A1R).

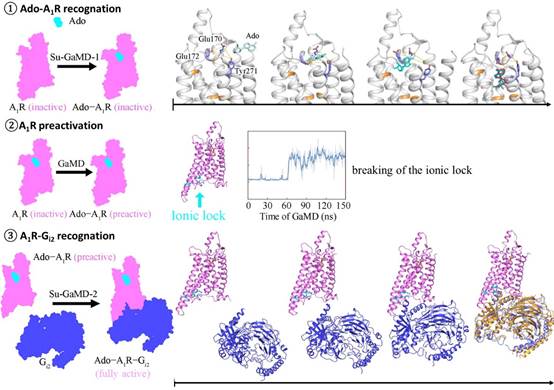
The research team incorporated a tabu-like supervision algorithm into the standard Gaussian accelerated molecular dynamics (GaMD) simulation and developed a new MD method, supervised Gaussian accelerated MD (Su-GaMD), which can reveal the recognition process of ligand-protein and protein-protein interactions on the nanosecond (ns) timescale. The simulation results showed that the full activation of A1R included Ado-A1R recognition, A1R pre-activation, and A1R-Gi2 recognition. The recognition of Ado underwent several different intermediate states, during which the residues, such as Glu170ECL2, Phe171ECL2, Glu172ECL2, Asn2546.55, Thr2576.58, Lys265ECL3, Tyr2717.36, and Thr2777.42, were important to the binding of Ado, which has been demonstrated in previous experiments. Then, the binding of Ado initiated conformational changes in the intracellular side of A1R, breaking the ionic lock between Arg1053.50/Arg1083.53 and Glu2296.30 and leading to the pre-activation of A1R. Finally, Gi2 gradually approached A1R at the intracellular side and bound with A1R through ICLs (especially ICL2), forming a complex of Ado-A1R-Gi2 (Ado RMSD = 2.0 Å, A1R RMSD = 1.7 Å, Gαi RMSD = 2.9 Å), which was consistent with the structure in previous experiments. The binding of Gi2 to the intracellular side of A1R reduced the volume of the extracellular Ado binding pocket and enhanced the binding ability of Ado to A1R, which was consistent with the previous experimental phenomenon of the beta-1 adrenergic receptor.
The study simulated the whole dynamic process from free Ado, apo-A1R, Gi2, to Ado-A1R-Gi2 complex in hundreds of ns simulations and revealed the mechanism of full activation of A1R, which is essential for understanding the signal transduction mechanism of G protein to G protein–coupled receptor (GPCR). The intermediate states revealed in the recognition of Ado-Gi2 and Gi2-A1R expand the new perspective in A1R drug design and give new ideas for R&D of A1R-targeting drugs.
Li Yang and Sun Jixue, doctoral students at the College of Pharmacy, Nankai University, are the co-first authors of this paper, and Associate Professor Li Dongmei and Professor Lin Jianping are the co-corresponding authors.
Paper link: https://www.pnas.org/doi/10.1073/pnas.2203702119